Looks good. Let's now write our function get_target_oligo(). This function has been through a few rounds of development in this notebook, so it has grown a bit complicated. Let's break down the steps required:
- Calculate homology arm length (from total
homology / 2), which assumes symmetric arms and an even homology length.
- Get the '+' strand sequence for the left and right arms. Note that this must be indexed properly, such that the left position is the last nt that is kept both in the genome and on the oligo (before attB), and the right position is the first nt after attB/in the genome.
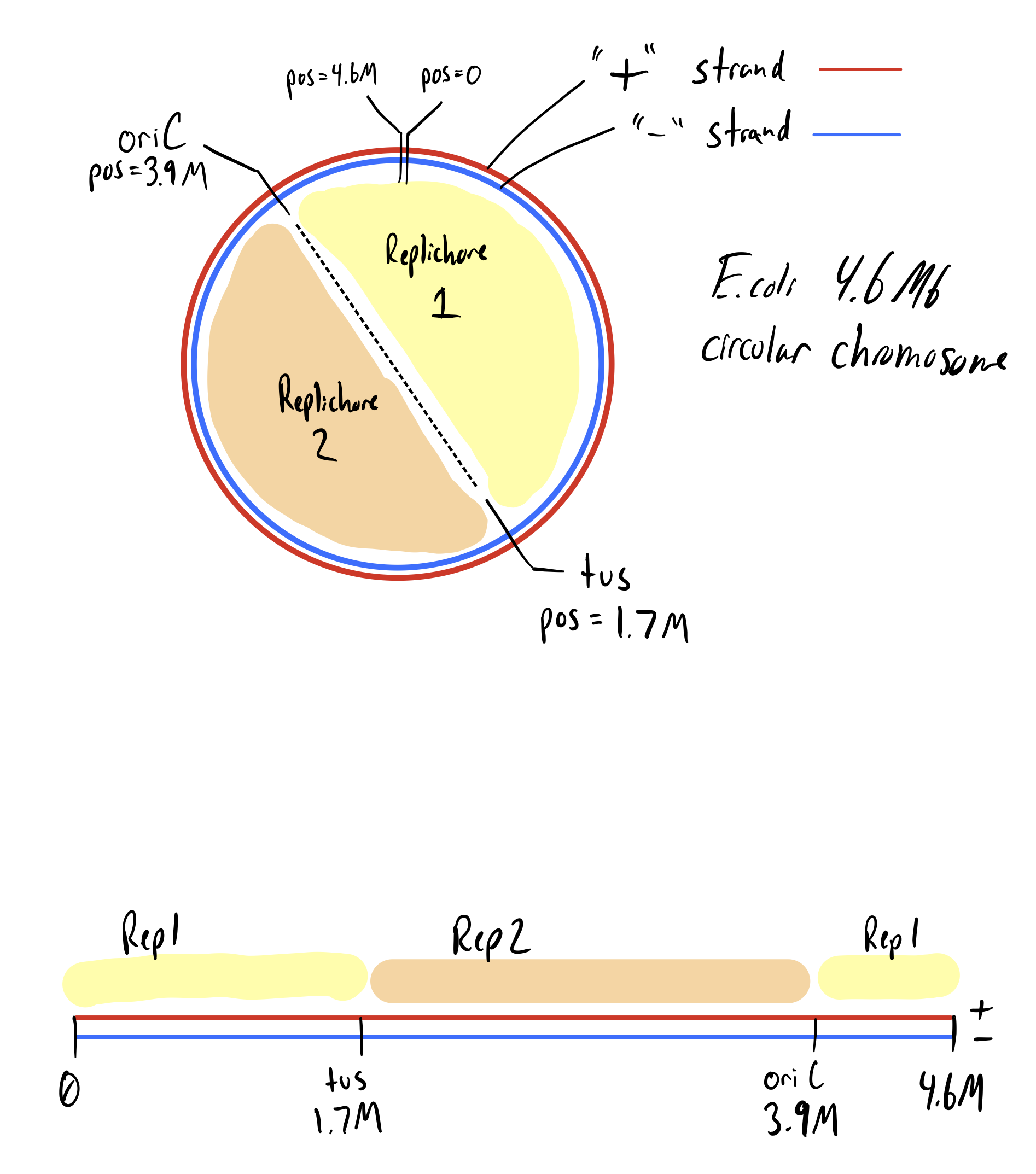
- Determine the replichore and reverse complement homology arms accordingly.
- Determine the direction of the attB site
This last part is somewhat complex, because there are 4 possible scenarios
Replichore = 1, Direction = '+'Replichore = 1, Direction = '-'Replichore = 2, Direction = '+'Replichore = 2, Direction = '-'
Let's start by explaining the simplest example first, Replichore = 2, Direction = '+'. In this case the homology arm sequences come directly from the '+' strand. If we are deleting a gene, then the 5' end of the oligo will have the left and upstream homology arm. Then the 3' end of the oligo will have the right and downstream homology arm. In this case we simply want to paste the fwd sequence of attB between these two arms to get an insertion of pInt in the expected direction (e.g. for pDel gro promoter facing downstream).
For Replichore = 2, Direction = '-', the gene is facing the opposite direction. Therefore the oligo still comes from the '+' strand, but now the 5' end of the oligo is the left and downstream homology arm and the 3' end is the right and upstream homology arm. Therefore we need to reverse complement the attB sequence, so that it is now facing downstream.
For Replichore = 1, Direction = '+', the gene is on the '+' strand, but the oligo sequence comes from the '-' strand. Therefore the 5' end of the oligo is right and downstream and the 3' end of the oligo is left and upstream. So, to get attB facing downstream, we need to reverse complement.
Finally for Replichore = 1, Direction = '-' we have the actual orientation / position of galK, so just think about that. The gene is on the '-' strand, but the oligo sequence also comes from the '-' strand. Here the 5' end of the oligo right and upstream and the 3' end is left and downstream. Therefore the forward attB sequence can be used to face downstream.
Here's a table to simplify things, and just remember that on the oligo, typically we want attB facing the same way as the gene: downstream.
| Replichore |
gene dir |
5' abs-rel pos |
3' abs-rel pos |
attB |
| 1 |
+ |
right-down |
left-up |
rev |
| 1 |
- |
right-up |
left-down |
fwd |
| 2 |
+ |
left-up |
right-down |
fwd |
| 2 |
- |
left-down |
right-up |
rev |
for + gene_dir locus should look like: (left) | upstream | attB_fwd | downstream (right)
for - gene_dir locus should look like: (left) | downstream | attB_rev | upstream (right)
With all of that reasoned out, we can write our function with some simple if statements: